Integrated Platform
The five modules of the BioPharmics Surflex Platform (Tools, Similarity, Docking, xGen, and Affinity) are fully integrated. The full software bundle provides a comprehensive predictive modeling workflow:
- 2D to 3D molecular conversion, with accurate chirality interpretation and enumeration facilities
- Conformer elaboration including complex macrocycles, also supporting the use of NMR restraints
- Protein structure preparation and alignment
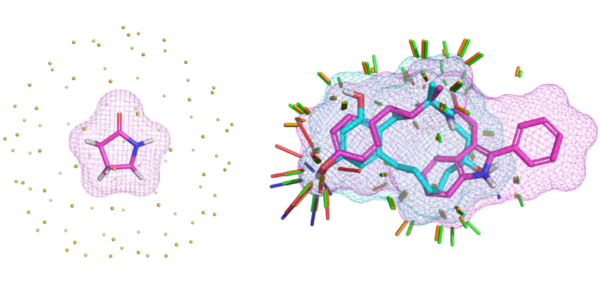
- Docking for pose prediction or virtual screening
- Real-space modeling of ligands as conformational ensembles within X-ray density maps
Software is available for Windows, Linux, and Mac platforms, with easy deployment across on-premises workstations and laptops as well as cloud-based computing resources.
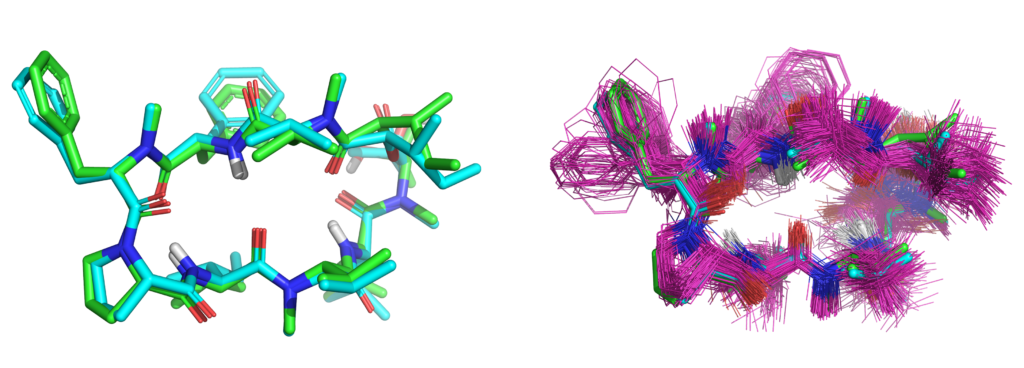

What’s New?
New Web Site!
June 1. 2024
Following our acquisition last year by by Optibrium, we have transitioned our web presence. This site will redirect to the new site soon. The new site can be found here.
Optibrium and BioPharmics Merge
August 31, 2023
We are delighted to announce that BioPharmics LLC is now the BioPharmics Division of Optibrium Ltd. See the press release for additional details. We will be continuing our work with a bigger and stronger team!
Version 5.193 Surflex Platform Released
October 16, 2024
This is a minor release (with a full example set): Release Notes. The release includes minor bug fixes and some improvements. See the Downloads page for details.
JCAMD: Macrocycle Optimization
February 26, 2024
We have published a study, with our colleagues from Corteva, demonstrating active-learning with QuanSA for lead-optimization of a macrocyclic natural product. This is in addition to our recent comprehensive study of complex macrocycle optimization through careful application of NMR restraints combined with molecular docking, molecular similarity, and estimation of bound ligand strain with colleagues from BMS.
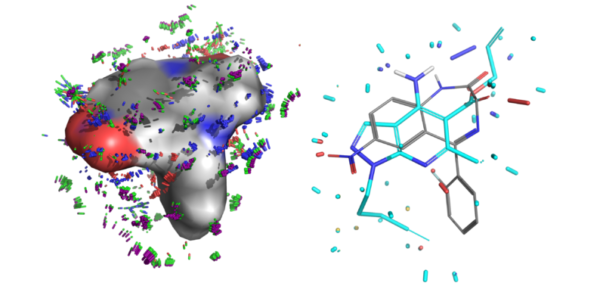
JMC: Strain/xGen Papers
January 27, 2023
We have published a comprehensive study of bound ligand strain with colleagues from Merck and BMS. This adds to a paper looking at peptide macrocycle strain energetics in the context of real-space refined ligand conformational ensembles. Both studies build on the xGen methodology where we showed that conformational ensembles, without atom-specific B-factors, are better models for ligands in terms of both fit to X-ray density and strain energy.
JCAMD: Cross-Docking of Macrocycles
October 16, 2024
We have extended our PINC cross-docking benchmark study to macrocyclic ligands. Prospective ensemble-docking performance for complex small molecules and macrocycles reached roughly 80% correct for the top two Surflex-Dock predicted docking solutions. The paper reports significantly better performance for the ForceGen/Surflex-Dock approach compared to widely used alternatives.